Amplicons

Get fast assemblies with our standard workflow, or sequence deeply with our End-to-End workflow.
Data and deliverables
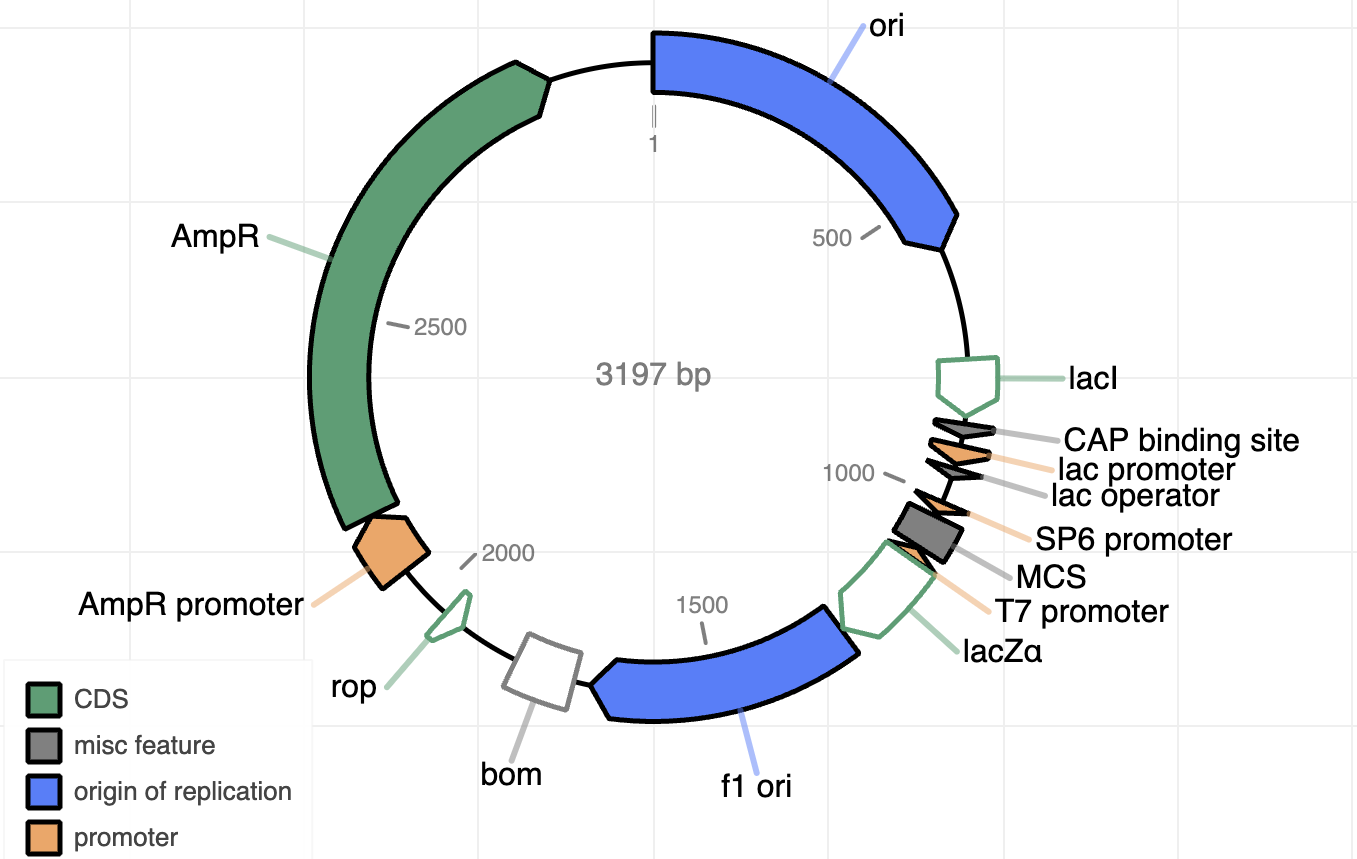
- Consensus sequence (.fasta)
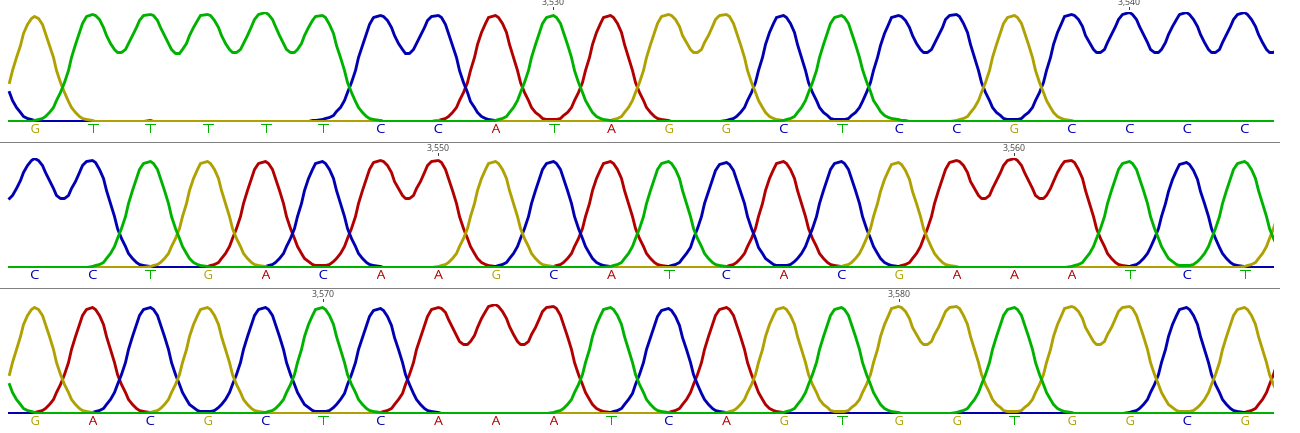
- Chromatogram (.ab1)
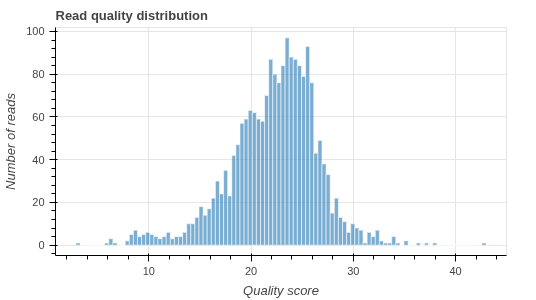
- Data quality report (.html)
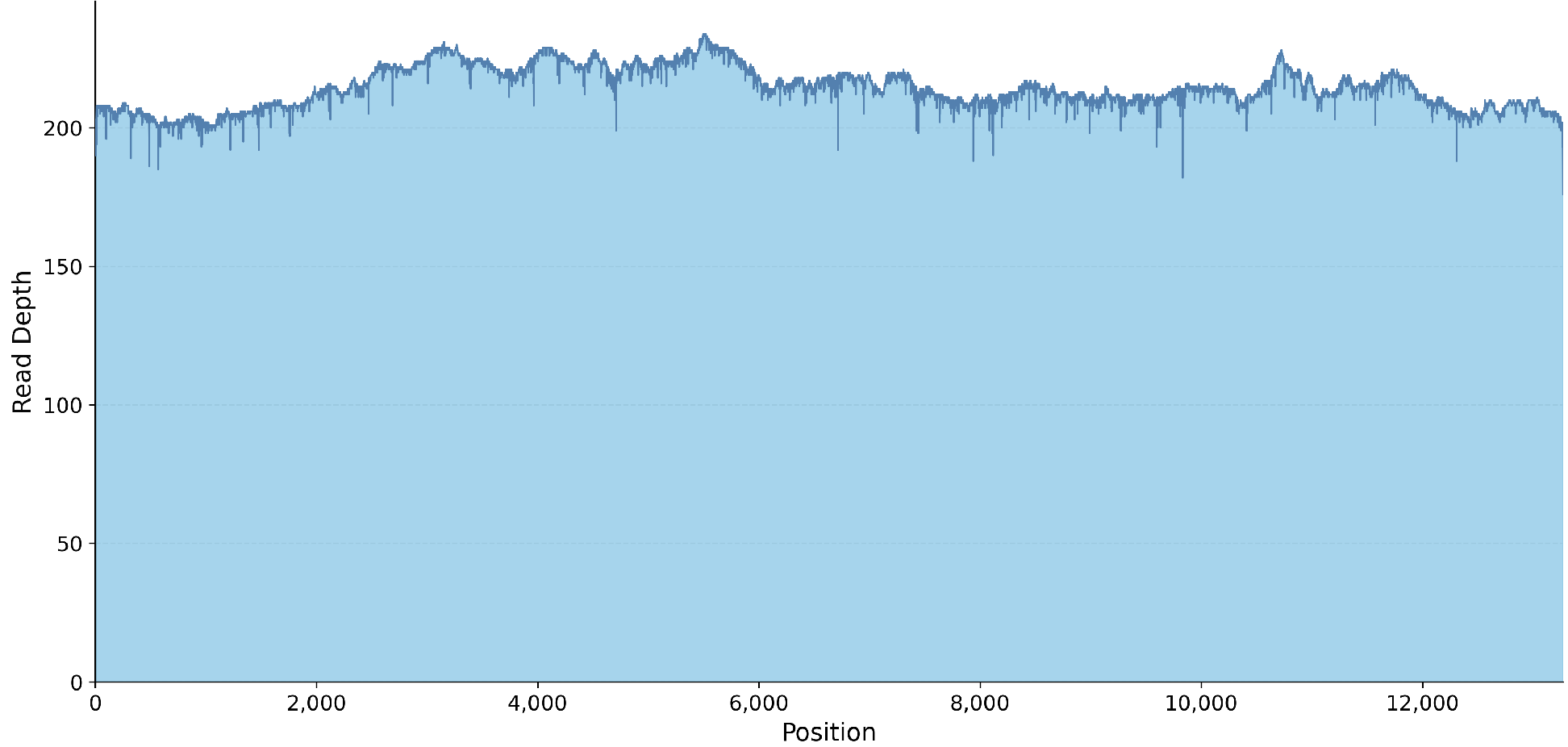
- Coverage plot (.png)
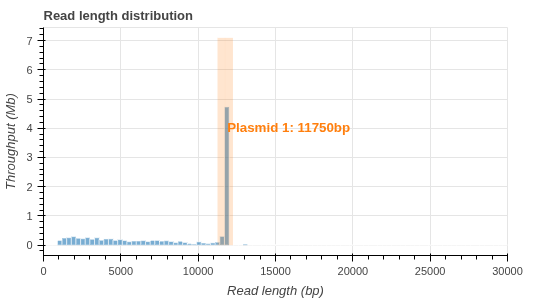
- Read length histogram (.png)
- Raw sequencing reads (.fastq)
- Per-base sequencing information (.txt)
- Responsive customer support

Pricing
All prices shown are Canadian dollars and subject to local sales tax.
Standard amplicons
Ideal for sequencing clonal PCR products larger than 500 bps. Double-stranded DNA fragments are tagmented during library preperation.
End-to-End Amplicons
Ideal for sequencing PCR products as small as 100 bps. Double-stranded DNA fragments are not tagmented during library preparation, resulting in full-length amplicon reads. Ideal for sequencing mixed populations.
Frequently Asked Questions
What's the difference between Standard and End-to-End Amplicons?
Standard Amplicons uses tagmentation during library prep, which fragments your amplicons before sequencing. This works well for clonal PCR products larger than 500 bp where you want fast turnaround and don’t need to preserve full-length reads.
End-to-End Amplicons skips tagmentation, producing full-length reads that span your entire amplicon. This is essential for amplicons under 500 bp, mixed populations, or any case where you need to see every read as a complete, unbroken sequence. Use our new Genotyping workflow for haplotype and variant analysis!
What sample types do you accept?
Purified PCR products from 500 bp to 25 kb for standard Amplicons, or > 100 bp for End-to-End Amplicons. We accept column-purified, bead-purified, or gel-extracted amplicons. Concentration requirements vary by service: Standard accepts 20–200 ng/µL, while End-to-End requires roughly 1 ng/µL per 100 bp of amplicon length. See the pricing tables above for exact specifications.
What files will I receive?
By default, you will receive a consensus sequence (.fasta), annotations (multiple formats), .fastq files, and quality control files (.png, .html). If you require custom analysis, please specify in the “Additional Information” section.
When will I receive my data?
Standard Amplicons complete in 1 business day from receipt of samples, although data is typically returned in hours after arrival. End-to-End Amplicons complete in 2 business days. If you need data ASAP, let us know.